INTRODUCTION
Inflammatory pain in mammalian sensory systems generated by intrinsic or extrinsic causes may be accompanied by physical damage but is frequently caused by abnormal firing of neurons or nerves without tissue damage. In neurophysiological mechanisms to process various pains that are induced by chemicals, physical stimulations, or biological factors, there are unique nociceptors or polymodal receptors that respond to a specific stimulus or multiple stimuli, respectively [1,2]. Accordingly, pain perception is carried out in the central nervous system, while the type of pain is determined peripherally [3,4]. Physical damage to tissues and nociceptive responses triggered by noxious stimuli or chemical toxins can activate intracellular signaling cascades to generate secondary pain responses, which tend to last longer than the primary pain responses [5,6]. The induction of intense pain in peripheral nerve endings occasionally tends to activate intracellular signaling in primary afferent neurons, resulting in hyperalgesia, allodynia, or neuropathic pain [7-9]. This secondary modulation of pain seems to be triggered by cellular mechanisms mediated by Ca2+ ions and reactive oxygen species (ROS). In primary afferent neurons of the mammalian somatosensory system, certain types of membrane proteins acting as ion channels or receptors simultaneously contribute to both the primary perception and secondary modulation of pain. In particular, transient receptor potential (TRP) receptors play important role in both the peripheral and central terminals of primary afferent neurons in recognizing and modulating nociceptive information [10,11].
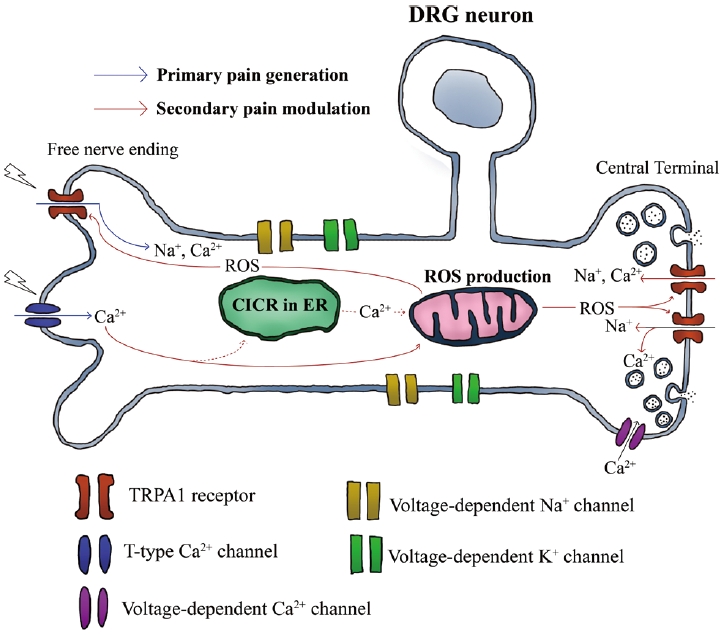
Recently, we reported that TRP ankyrin 1 (TRPA1) receptors contributed to prolonged pain responses in rats, which were induced by jellyfish toxin [12]. In that study, the contribution of TRPA1 but not TRP vanilloid 1 (TRPV1) was crucially involved in the secondary pain modulation triggered by ROS and Ca2+ influx through T-type Ca2+ channels in dorsal root ganglion (DRG) neurons. In this paper, we briefly review the functions of TRPA1 receptors that interact with intracellular signaling during pain modulation. TRPA1 receptors are major polymodal nociceptors that generate primary pain responses in the peripheral nerve endings of the primary afferent neurons. In addition, these receptors play an important role in generating synaptic depolarization by ROS signaling in the central terminal of primary afferent neurons, regulating long-lasting pain responses. Therefore, it is important to know the functional characteristics of the TRPA1 receptor rather than other types of TRP receptors to understand secondary pain modulation in sensory neurons under several pathogenic conditions such as neuropathic pain syndromes and autoimmune diseases, which are mediated by oxidative signaling and cause chronic pain.
DISTRIBUTION OF TRPA1 RECEPTORS
TRPA1 receptors, similar to other cation-permeable membrane proteins, are expressed in various mammalian tissues. These receptors are dominantly found in gastro-intestinal organs and skeletal systems such as small intestine, lung, pancreas, and skeletal muscles [13]. In the somatosensory system, TRPA1 receptors typically express in basal keratinocytes as a polymodal nociceptor responsive to physical, thermal, and chemical stimulations.14 Furthermore, these receptors regulate inflammatory gene expression in keratinocytes, monocytes, and macrophages, promoting the expression of interleukin (IL)-1α and IL-1β that may enhance cutaneous neurogenic inflammation directly or indirectly [14]. A variety of senses originated from skin are transferred to the spinal cord though the primary afferent neurons (i.e., DRG neurons) which express large amount of TRPA1 receptors in their fibers as well as a soma [15]. In a mammalian nervous system, non-neuronal cells such as astrocytes, peripheral oligodendrocytes, and Schwann cells also express large amount of TRPA1 receptors [16,17]. Therefore, the distribution of these receptors indicates their role as a primary nociceptor to generate pain against dynamic extrinsic and intrinsic stimuli in the whole body.
DYNAMIC PAIN MODULATION OF TRPA1 RECEPTORS
Nociceptive responses mediated by TRPA1 receptors are triggered by several kinds of extrinsic and intrinsic stimulatory factors that induce cationic currents through the receptors. Early studies on the physiological functions of TRPA1 receptors focused on their ability to detect cold-related thermal pain, but later studies proposed that they might modulate the nerve ending sensitivity to cold stimulation as well as thermal nociception [15,18]. Furthermore, various chemicals have been reported to directly or indirectly activate TRPA1 receptors. Endogenous or exogenous chemicals that activate these receptors include allyl isothiocyanate, cinnamaldehyde, formaldehyde, acetaldehyde, 4-hydroxynonenal, ROS, 12-lipoxygenase-derived hepoxilin A3, and 5-6-epoxyeicosatrienoic acid [19,20]. Chemoreception to multiple stimuli by TRPA1 receptors is based on their dynamic binding properties inside and outside the neuronal membrane. For example, TRPA1 receptors are well known to covalently interact with a number of agonists through their N-terminal region containing cysteine and lysine residues to activate themselves [21]. Hypoxia also indirectly activates TRPA1 receptors through prolyl hydroxylase (PHD)-mediated signaling, because hypoxia-inhibited PHD elicits the PHD-dependent hydroxylation of a proline residue existing within the N-terminal ankyrin repeat domain, enhancing the sensitization of TRPA1 receptors to ROS [22,23].
TRPA1 receptors also contribute to secondary pain modulation, correlating with inflammatory pain, hyperalgesia, and allodynia, through specific receptor-dependent signaling pathways to upregulate receptor activity. Various inflammatory mediators such as growth factors, bradykinins, protease, and thymic stromal lymphopoietin, known as an epithelial-derived cytokine, have been reported to prevent desensitization of TRPA1 receptors [24-26]. The activation of TRPA1 receptors increases intracellular Ca2+ levels by inducing Ca2+ influx and Ca2+ outflux from the endoplasmic reticulum in primary afferent neurons to propagate nociceptive signals to the central nervous system [27,28]. This seems to directly affect the release of neurotransmitters involved in pain generation and modulation, such as substance P, calcitonin gene-related peptide (CGRP), and glutamate, amplifying excitatory postsynaptic potentials in the spinal pain pathway [29,30].
The TRPA1 receptor also acts as a secondary pain modulator that is dependent on Ca2+ ions. Recently, jellyfish venom extracted from Chrysaora pacifica was reported to activate T-type Ca2+ channels and prolong the pain responses in rats [12]. In that study, the long-lasting pain was dependent on TRPA1 receptors and ROS, indicating that ROS, which was increased by Ca2+ influx through T-type Ca2+ channels, might intracellularly activate TRPA1 receptors and induce persistent pain responses (Fig. 1). Therefore, TRPA1 receptors are targeted by Ca2+-dependent signaling during secondary pain modulation.
It is well known that intracellular Ca2+ regulates TRPA1 receptors. The N-terminal domains of TRPA1 receptors have a binding site for Ca2+ ion to activate receptors [31,32]. Although there are many types of Ca2+ channels that induce Ca2+ influx, T-type channels seem to be closely correlated with the function of TRPA1 receptors. This type of Ca2+ channels showing a low voltage-activated property is primarily involved in pain modulation [33]. Also, they are abundantly expressed in nociceptive sensory neurons, enhancing the firing rate of action potentials, neuronal excitability, and pain transmission [33-35]. Particularly, Cav 3.2 channel, a T-type channel subtype, tended to be upregulated by inflammation, was increasingly expressed during the induction of neuropathic pain in animal models [36,37]. In another study, only verapamil used to block T-type channels but not nimodipine, significantly suppressed TRPA1-mediated pain and ROS production in primary afferent neurons. This provides clear evidence that T-type Ca2+ channels, but not L-type channels, contribute to the activation of TRPA1 receptors in primary somatosensory neurons, indicating a specific correlation between them in pain modulation [12].
ROS AND TRPA1
TRPA1 receptors also play a critical role in pain modulation during tissue injury and inflammation that activate oxidative signaling via dynamic mechanisms [38-40]. A number of types of mammalian eukaryotic cells can produce ROS via the aerobic metabolism. Phagocytes such as polymorphonuclear cells, macrophages, and non-professional phagocytes including endothelium, epithelium, fibroblasts, and smooth muscle cells, which express nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), can physiologically and pathologically produce ROS [41]. Interestingly, the TRPA1 receptor is well known to be targeted by various types of ROS such as hydrogen peroxide (H2O2), hydroxyl radical (OH-), and hypochlorite (OCl-) [42,43]. TRPA1 receptors act as sensors to detect ROS in epithelial cells of the skin, respiratory, and gastrointestinal systems [44].
In the nervous system, TRPA1 receptors can be activated by ROS via dependent or independent pathways for pain generation [45,46]. In ROS-dependent pathways, TRPA1 receptors can be activated in both hyperoxic and hypoxic states. These receptors tend to be activated by a low redox potential threshold and are hypersensitive to electrophilic compounds [47]. The covalent modification of cysteine by hyperoxia can activate TRPA1 receptors, and the mutation of cysteine Cys633Ser or Cys856Ser deactivates them. In hypoxia, TRPA1 receptors can be activated by the inactivation of oxygen-sensitive PHD, which generally suppresses TRPA receptors in normoxia [48]. Recently, it has been reported that CGRP contributes to the intracellular interaction of TRPA1, and ROS is significantly important for regulating cortical susceptibility to spreading cortical depression [49]. In that study, ROS seemed to activate TRPA1 receptors expressed in both neurons and glia and enhanced CGRP production in the central nervous system. These results indicate that the TRPA1 receptor acts as a cation channel to participate in the dynamic metabolic processes of neurons in an ROS-dependent manner.
Oxidative signaling cascades of sensory neurons are involved in the protective mechanisms that are activated by various adverse events. Chemotherapeutic-induced peripheral neuropathy (CIPN), which causes paresthesia, spontaneous pain, hyperalgesia, and cold hypersensitivity, has been reported to trigger oxidative stress in mammalian neurons [50]. In a mouse model, CIPN was induced by oxaliplatin and its metabolite oxalate, and then directly inhibited PHD activity, enhancing cold hypersensitivity of TRPA1 receptors [23,51]. Therefore, it is also possible that, in some cases, the activation of TRPA1 during oxidative stress does not require ROS. However, the correlation between ROS generation and TRPA1 receptor activation appears to be more evident in pain modulation. In 2013, Nesuashvilie et al. [52] reported that acute mitochondrial dysfunction in bronchopulmonary C-fibers of mice, which was induced by antimycin A, enhanced ROS production and activated TRPA1 receptors. However, it was recently confirmed that antimycin-A required Ca2+ influx and ROS generation to activate TRPA1 receptors in nociceptive neurons [53]. Therefore, for pain modulation, ROS are necessary and sufficient for enhancing nociceptive responses of TRPA1 receptors via activating Ca2+ signaling.
The regulation of eukaryotic inflammation provides additional evidence of the correlation between TRPA1 receptors and ROS signaling. In lung epithelial cells and nociceptors expressing TRPA1 receptors, lipopoly-saccharide seems to directly activate TRPA1 receptors, resulting in the increase of intracellular Ca2+ and subsequent activation of NADPH oxidase, which enhances the production of ROS. This signal cascade finally reaches the activation of mitogen-activated protein kinase (MAPK) and nuclear factor-κB (NF-κB) signaling for the production of pro-inflammatory cytokines [54,55]. Therefore, the functional coupling of TRPA1 receptors and ROS not only results from oxidative signaling triggered under various abnormal conditions, but also actively plays an important role to regulate ROS-mediated inflammatory signaling cascades.
CONCLUSION
Although many researchers have focused on the function of TRPA1 receptors as thermoreceptors and polymodal nociceptors responsible for external stimuli, these receptors also play a role in generating intrinsic signals by leading to membrane depolarization, which contributes to secondary pain modulation or oxidative signaling. Furthermore, their participation in intracellular communication is crucial in identifying clinical problems by inducing nociceptive responses in autoimmune diseases, neuropathic pain syndromes, and oxidation-mediated metabolic diseases. Therefore, it is important to study the kinetics and physiological roles of TRPA1 receptors as the dominant factors in intrinsic signal modulation.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print